1月22日,我校基础医学院侯宇教授、赵雪雅副教授团队联合附属第二医院血液内科邓建川教授团队在国际知名期刊Journal of Experimental Medicine发表了题为Inhibition of DEK restores hematopoietic stem cell function in Fanconi anemia的研究论文。

范可尼贫血(Fanconi anemia,FA)是一种累及多个系统的常染色体隐性遗传性疾病,骨髓造血功能衰竭是FA的典型临床表现。造血干细胞(Hematopoietic stem cells, HSCs)是血液系统中具有长期自我更新能力和多向分化潜能的成体干细胞,HSCs耗竭是FA骨髓衰竭的关键原因。研究发现范可尼基因突变导致HSCs对复制压力更为敏感,造成其功能受损及数量耗竭,但相关机理尚不清楚。因此,深入研究范可尼基因突变导致HSCs耗竭的具体机制对理解FA发生发展及临床干预具有重要意义。
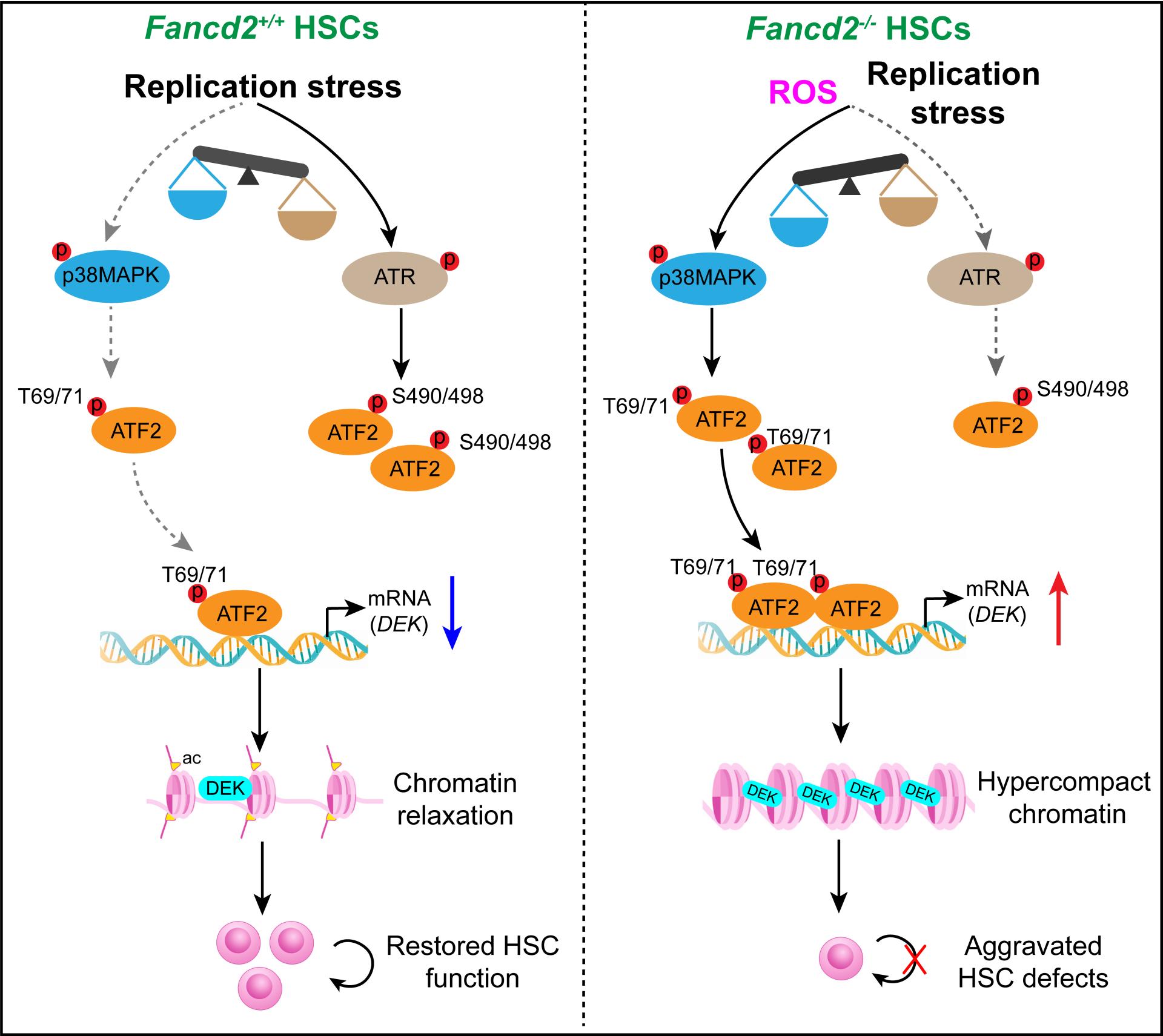
在本研究中,研究人员利用多组学技术(RNA-seq,ATAC-seq及CUT&Tag-seq)绘制了复制压力下HSCs的响应图谱,首次发现HSCs在复制压力下适应性出现H3K27ac修饰及染色质可及性增加现象。相反限制染色质的松弛会阻碍复制压力的消减,更会直接引起复制压力,进而损害HSCs的维持及造血功能。研究人员发现FA患者中骨髓CD34+细胞染色质可及性较正常样本显著降低,揭示了FA患者造血干祖细胞中复制压力消减受阻的表观机制。进一步的机制研究发现染色质架构蛋白DEK是关键效应因子,其是限制H3K27ac修饰及染色质可及性的关键蛋白。复制压力下,野生型小鼠的HSCs通过ATR激酶促进转录因子ATF2的S490/498位点磷酸化,造成ATF2与DEK基因启动子区域结合减少,进而抑制DEK基因转录,最终促进染色质松弛;相反范可尼贫血模型小鼠(Fancd2敲除小鼠,Fancd2-/-)的HSCs因p38激酶过度激活,促进ATF2的T69/71位点磷酸化,维持高转录活性,导致DEK异常高表达,进而阻碍染色质松弛 并加剧复制压力。此外研究人员证实FA患者骨髓CD34+细胞中DEK异常高表达,敲低DEK或者利用DEK抑制剂(核酸适配体DTA-64)处理都能有效促进FA患者来源的CD34+细胞在体造血功能增强。这些发现为解释FA中HSCs功能缺陷提供了新见解,更为干预FA提供了新靶点。
我校基础医学院侯宇教授,赵雪雅副教授,以及附属第二医院血液内科邓建川教授为该论文的共同通讯作者,我校基础医学院陈哲副研究员、博士研究生伍凤、博士后李言及博士研究生李蕾是论文的共同第一作者。该项目得到了国家自然科学基金、重庆市杰出青年科学基金、重庆市自然科学基金、博鱼平台重大科技人才项目A类等项目的资助。
全文链接:
https://rupress.org/jem/article-abstract/222/3/e20241248/277224/Inhibition-of-DEK-restores-hematopoietic-stem-cell?redirectedFrom=fulltext
 电话:023-68485000/68485111
电话:023-68485000/68485111
 地址:重庆市渝中区医学院路1号
地址:重庆市渝中区医学院路1号
 邮编:400016
邮编:400016
